Версия для скачки/печати: Скачать
 * формат .doc
* формат .doc
Страница 1
Пренатальная патология
Актуальность темы
Прежде чем приступить к изучению темы настоящей лекции необходимо вспомнить некоторые общие философские понятия из курса биологии и гистологии, в частности, что такое онто - и филогенез, а также этапы нормального развития плода.
Онтогенез – это процесс индивидуального развития организма от момента его зарождения до смерти.
Термин "онтогенез” введен известным немецким ученым Э.Геккелем в связи с формулировкой основного биологического закона, согласно которому онтогенез есть краткое и сжатое повторение филогенеза – процесса исторического развития органического мира (отдельных типов, классов, отрядов, семейств, родов, видов живых организмов).
В основе онтогенеза лежит цепь строго определенных последовательных биохимических, физиологических и морфологических изменений, специфических для каждого из периодов индивидуального развития организма.
В соответствии с этими изменениями онтогенез принято делить на эмбриональный (зародышевый, или пренатальный) период и постэмбриональный (послезародышевый, или постнатальный). Первый охватывает время от оплодотворения до рождения, второй – от рождения до смерти.
Общепризнано, что организмы, размножающиеся половым путем, зарождаются в момент оплодотворения яйцеклетки сперматозоидами, в результате чего образуется зародыш. Некоторые исследователи выделяют еще предзародышевую стадию онтогенеза, соответствующую формированию яйцеклетки и сперматозоидов, которая во многом определяет последующее развитие зародыша.
В результате первоначального деления оплодотворенной яйцеклетки (зиготы) возникает клеточная масса (бластоциста), состоящая из внутренней клеточной массы (эмбриональный диск), окруженной наружным слоем (трофобластом). Клетки трофобласта активно пенетрируют эндометрий и формируют плаценту. Клетки эмбрионального диска являются тотипотентными (то есть, обладают способностью дифференцироваться в любой тип клеток организма). В результате первоначальной дифференцировки образуются три первичных зародышевых листка: эктодерма, мезодерма, энтодерма, из которых затем образуются различные органы организма в результате деления, организации и дифференцировки составляющих их клеток. Скопление примитивных клеток, из которых в дальнейшем развивается орган, названо закладкой органа. Полностью развитый орган составлен из высокодифференцированных клеток, выполняющих специфическую функцию и имеющих ограниченную способность к делению. Большинство органов человека окончательно формируется и функционирует уже к моменту рождения. Одни органы, такие как сердце, полностью формируются еще во внутриутробной жизни, а другие, например, легкие, продолжают развиваться еще в течение 3-4 недель после рождения. Мозг продолжает развиваться после рождения, достигая зрелости к семилетнему возрасту. Половое развитие происходит во время пубертатного периода.
Понятие "пренатальная (антенатальная) патология” включает в себя все патологические процессы и состояния человеческого зародыша от момента оплодотворения и до рождения ребенка. Пренатальный период человека исчисляется 280 днями, или 40 неделями, после чего наступают роды.
Все развитие, начиная от созревания половой клетки (гаметы) до рождения зрелого плода, делят на два периода:
период прогенеза;
период киматогенеза (от греч. kyema — зародыш).
Период прогенеза соответствует созреванию гамет (яйцеклеток и сперматозоидов) до оплодотворения. В этот период возможно возникновение патологии гамет — гаметопатии.
Период киматогенеза — соответствует периоду от оплодотворения до родов. В нем различают три периода:
а) бластогенез — период от оплодотворения до 15 дня беременности. В этот период идет дробление яйца, заканчивается образованием эмбриобласта и трофобласта;
б) эмбриогенез — период с 16 дня до 75 дня беременности, идет основной органогенез и образуется амнион и хорион;
в) фетогенез — период с 76 дня по 280 день беременности, происходит дифференцировка и созревание тканей плода, образование плаценты, а также рождение плода. Фетогенез в свою очередь делится на:
ранний фетальный период (76-180 день беременности);
поздний фетальный период (181-280 день беременности).
К концу раннего фетального периода незрелый плод приобретает жизнеспособность. Поздний фетальный период завершается созреванием плода с одновременным старением плаценты. С периодом киматогенеза совпадает период киматопатии.
Последовательность появления и развития различных признаков организма человека происходит под контролем генетического аппарата. Онтогенез, таким образом, является процессом реализации полученной от родителей наследственной информации в конкретных условиях окружающей среды. Вместе с тем следует подчеркнуть, что наряду с жестко запрограммированностью "работы” генов, всего генетического аппарата окружающая среда в процессе онтогенеза оказывает разнообразное воздействие на активность генов.
Изучение генетической программы онтогенеза в значительной степени помогло выявлению причин, а также разработке методов лечения заболеваний, связанных с нарушением деятельности генетического аппарата.
Знание особенностей течения таких заболеваний, причин их появления повышает шанс на излечивание. Эти данные необходимы для медико-генетического конультирования при планировании семьи, поскольку выявление носительства генов, обусловливающих развитие той или иной болезни, позволяет врачу дать рекомендации о степени риска передачи таких генов потомству.
Цель обучения – уметь определять морфологические проявления пренатальной (антенатальной) патологии, объяснить их причины и механизм развития, оценить исход и определить значение вероятных осложнений для организма.
Для чего необходимо уметь:
определить видимые макро- и микроскопически признаки гаметопатий, объяснить причины, механизм развития, исход и оценить их значение;
определить видимые макро- и микроскопически признаки киматопатий, объяснить причины, механизм развития, исход и оценить их значение;
определять морфологические проявления важнейших врожденных пороков развития, объяснить причины, механизм развития, исход и оценить их значение.
ГАМЕТОПАТИИ
Понятие "гаметопатии” охватывает все виды повреждений мужской и женской гаметы (яйцеклетки и сперматозоида), возникающие во время ово- и сперматогенеза до оплодотворения. Гаметопатии обусловлены, главным образом, мутациями. Термин "мутации” широко используется для обозначения любых стабильных врожденных генетических изменений, несмотря на то, связаны они или нет с определяемыми структурными нарушениями в хромосомах. В зависимости от того, в каких структурах наследственного аппарата гаметы произошла мутация возможно развитие различных мутаций: генных, хромосомных или геномных.
Нормальный хромосомный набор: Нормальные клетки человека имеют 46 хромосом: 22 пары аутосом и две половые хромосомы. По одной хромосоме в каждой паре человек получает от каждого родителя.
Половые хромосомы — это пара Х хромосом у женщин и Х и Y хромосомы у мужчин. Генетический пол может быть установлен при исследовании кариотипа (этот метод является наиболее точным) или при исследовании клеток на наличие телец Барра. Когда в клетке присутствует две Х хромосомы (как у нормальной женщины), одна из них (тельце Барра) инактивируется и конденсируется на ядерной мембране. Отсутствие тельца Барра свидетельствует о наличии только одной Х хромосомы (у нормального мужчины (XY) и при синдроме Тернера (ХО)). Тельца Барра наиболее легко определяются в мазках многослойного эпителия, которые получают путем соскабливания буккальной слизистой оболочки. Y хромосому можно выявить в интерфазном ядре из-за сильной ее флуоресценции в ультрафиолетовом свете после предварительной окраски квинакрином. Данный метод является одним из способов определения генетического пола.
Гаметы являются носителями генов, унаследованных ими не только от родителей, но и от всех отдаленных предков. Тяжелые повреждения гамет могут вести к их гибели, развитию бесплодия, спонтанных абортов и выкидышей. Гамета с дефектом гена или генов может стать источником наследственных пороков развития или заболеваний.
НАРУШЕНИЯ РАЗВИТИЯ ПЛОДА В РЕЗУЛЬТАТЕ ГЕНЕТИЧЕСКИХ ПОВРЕЖДЕНИЙ. ХРОМОСОМНЫЕ АБЕРРАЦИИ.
Механизмы хромосомных аберраций:
Нерасхождение гомологичных хромосом происходит при первом мейотическом делении, в результате которого образуются гаметы. Таким образом, одна гамета получает пары хромосом, а вторая не получает ни одной. После второго мейотического деления образующиеся гаметы содержат 24 или 22 хромосомы. Такие гаметы называются анэуплоидными (количество хромосом в них не равно 23, нормальному гаплоидному набору человека). При оплодотворении анэуплоидной гаметы нормальной образуется анэуплоидная зигота, имеющая или три одинаковые хромосомы (трисомия) или только одну (моносомия). Трисомия или моносомия половых хромосом обычно совместима с жизнью, например, синдром Клайнфельтера (ХХY), синдром Тернера (ХО). При аутосомной моносомии теряется огромное количество генетического материала, поэтому она обычно летальна. Некоторые аутосомальные трисомии (21, 13 и 18) совместимы с жизнью, но связаны с тяжелыми нарушениями.
Нерасхождение хромосом на ранних этапах развития зиготы приводит и развитию мозаицизма: присутствию в организме человека двух или более генетически различных клеточных популяций. Фенотипические проявления нарушений колеблются от нормы к классическим проявлениям хромосомных аберраций. Так, например, у человека с мозаичным кариотипом Тернера (45,Х/46,ХХ) проявления будут колебаться от признаков нормальной женщины до проявлений классического синдрома Тернера (45,ХО).
Делеция — это потеря части хромосомы в результате ее разрыва. Большинство делеций летальны в результате потери огромной части генетического материала. Делеция короткого плеча 4 и 5 хромосом приводит к развитию хорошо изученных синдромов (синдром Вольфа и синдром кошачьего крика соответственно). Частичная делеция хромосом часто определяется в опухолевых клетках.
Транслокация — это перенос отдельного сегмента одной хромосомы в другую хромосому. При сбалансированной транслокации весь генетический материал сохраняется и остается функционально способным, поэтому фенотипических проявлений нет. Наиболее известной сбалансированной транслокацией является перенос целой 21-ой хромосомы в 14-ую хромосому. Такие люди имеют 45 хромосом с отсутствием одной 14 и одной 21 хромосомы и наличием ненормальной большой хромосомы, содержащей материал обоих хромосом (14 и 21); в основном поражаются мужчины, кариотип которых обозначается как 45,XY, t(14; 21). У таких людей могут формироваться аномальные гаметы, в результате чего у потомков может развиваться моносомия 21 (несовместимая с жизнью) или транслокационный тип синдрома Дауна. Другие типы сбалансированных транслокаций являются причиной привычных, или повторяющихся абортов.
Изменения в единичном нуклеотиде — проявляются нарушением расшифровки триплетного кода, но не вызывает определяемых структурных изменений в хромосомах. Эти нарушения названы болезнями единичного нуклеотида.
ПРИЧИНЫ ХРОМОСОМНЫХ АБЕРРАЦИЙ
Большинство хромосомных дефектов происходит "случайно”, то есть без видимой причины, но в некоторых случаях она может быть установлена:
Возраст матери — при увеличении возраста матери происходит учащение случаев нерасхождения хромосом, подтверждением чего является трисомия 21-ой хромосомы (синдром Дауна). Риск трисомии 21-ой хромосомы, который составляет 1:1500 живорожденных у женщин в возрасте моложе 30 лет, увеличивается до 1:30 у женщин старше 45 лет. Поэтому беременным женщинам старше 35 лет настоятельно рекомендуется производить хромосомный анализ клеток плода, полученных при амниоцентезе. Увеличение материнского возраста также связано с другими синдромами нерасхождения хромосом, например, синдром Кляйнфельтера.
Ионизирующее излучение — увеличение количества случаев хромосомных аберраций наблюдалось у людей, выживших после атомной бомбардировки Хиросимы и Нагасаки. Не существует "безопасной” дозы ионизирующей радиации, поэтому диагностическое рентген-исследование брюшной полости у беременных должно производиться только в исключительных случаях.
Медикаменты и другие химические вещества являются менее частой причиной структурных изменений хромосом. Использование противоопухолевых лекарств, которые нарушают синтез ДНК, в ранние сроки беременности могут привести к гибели плода. Множество широко используемых лекарств, в том числе и аспирин, вызывают изменения кариотипа в тканевых культурах; однако такой эффект in vivo не описан.
НАИБОЛЕЕ ЧАСТЫЕ АУТОСОМНЫЕ НАРУШЕНИЯ
Синдром Дауна является наиболее частым аутосомным нарушением (1:700 живорожденных). Он возникает в результате наличия третьей 21 хромосомы, что приводит к развитию характерных клинических проявлений. Дети имеют характерный косой разрез глаз с уплощенным профилем, кососмотрящие глаза, резко выраженные вертикальные кожные складки, прикрывающие медиальный угол глазной щели, так называемое сходство с лицами азиатов, раньше называемое "монголоидным”. Постоянным признаком является отставание в умственном развитии. 30% пациентов имеют врожденные пороки сердца, наиболее часто — дефект межжелудочковой перегородки. Также у этих больных повышена заболеваемость различными инфекциями, язвой двенадцатиперстной кишки и острой лейкемией. У пациентов, доживающих до взрослого возраста (50% и более) часто развивается пресенильная деменция с признаками болезни Альцгеймера. При этом в очагах повреждения накапливается бета-амилоид; предполагается, что ген бета-амилоида расположен в 21 хромосоме. Мужчины с синдромом Дауна обычно бесплодны, а женщины могут рожать детей. Потомки матерей с синдромом Дауна могут быть нормальными, потому что лишняя 21 хромосома содержится не во всех гаметах.
Синдром Эдварда — трисомия 18 хромосомы (47, ХХ/ХY, +18) встречается редко. Клинически он проявляется отставанием в физическом и умственном развитии, сопровождаемым характерными физическими недостатками, такими как "стопа рокера” и сжатые в кулаки руки с перекрещивающимися пальцами. В результате тяжелых поражений дети редко выживают более года.
Синдром Патау — трисомия 13 хромосомы (47, ХХ/ХY, +13) также встречается редко. Большинство детей умирает сразу после рождения. Трисомия 13 хромосомы характеризуется нарушением развития подкорковых структур мозга (отсутствие обонятельных луковиц, слияние лобных долей и единственный желудочек головного мозга) и срединных структур лица (расщепление губы, расщепление твердого неба, дефекты носа, единственный глаз [циклоп]).
Синдром кошачьего крика (сri du chat) — это нарушение вызвано делецией короткого плеча 5 хромосомы. Мяуканье и звуки, подобные кошачьему крику типичны для этой патологии. Часто наблюдается отставание в умственном развитии и пороки сердца. Срок жизни таких больных незначительно выше, чем у пациентов с трисомией 18 и 13 хромосом.
НАИБОЛЕЕ ЧАСТЫЕ АНОМАЛИИ ПОЛОВЫХ ХРОМОСОМ
Синдром Кляйнфельтера (тестикулярная дисгенезия) — встречается довольно часто, приблизительно 1:600 живорожденных мальчиков. Обычно причиной является нерасхождение Х-хромосом у матери ребенка, что проявляется наличием лишней Х-хромосомы (47, ХХY), реже больные с синдромом Кляйнфельтера могут иметь две и более лишних Х-хромосом (48, ХХХY или 49, ХХХХY). Y-хромосома контролирует дифференцировку яичек из примитивных закладок, в результате чего формируется мужской фенотип. До пубертатного периода никаких клинических проявлений не наблюдается. Лишняя Х-хромосома нарушает нормальное развитие яичек в пубертатном периоде неизвестным способом. Яички остаются маленькими и не продуцируют сперматозоиды, больные обычно бесплодны. Уровень тестостерона в крови низкий, что приводит к нарушению развития вторичных половых признаков. У больных имеется склонность к высокому росту (тестостерон ускоряет окостенение эпифизов) и евнухоидный внешний вид с высоким голосом, маленьким пенисом и ростом волос по женскому типу. Также иногда наблюдается гинекомастия. Иногда наблюдается снижение интеллекта. Диагноз синдрома Кляйнфельтера может быть установлен при нахождении телец Барра в соскобах буккального эпителия у лиц, имеющих мужской фенотип или при анализе кариотипа.
Синдром Тернера (яичниковая дисгенезия) — встречается довольно часто, приблизительно 1:2500 живорожденных девочек. Он развивается в результате расхождения Х-хромосом у любого из родителей, что приводит к отсутствию одной Х-хромосомы (45, ХО). Примерно у половины пациентов с синдромом Тернера обнаруживается мозаицизм (45, ХО/46, ХХ) в результате нерасхождения Х-хромосом на ранних стадиях развития эмбриона. В некоторых случаях вторая Х-хромосома присутствует, но в ней выявляются тяжелые нарушения (изохромосома, частичная делеция и др.).
Потеря второй Х-хромосомы обычно приводит к гибели плода. У выживших детей наблюдается лимфедема шеи, которая присутствует и у взрослых, приводя к формированию толстой шеи. Часто наблюдаются врожденные аномалии сердца (наиболее часто — коарктация аорты), низкий рост, ожирение и нарушения строения скелета. Интеллект не нарушен. В присутствии одной Х-хромосомы (и отсутствии Y-хромосомы) примитивные половые железы развиваются как яичники. Отсутствие второй Х-хромосомы приводит к нарушению развития яичников в пубертатном периоде. Яичники остаются маленькими и в них не обнаруживаются примордиальные фолликулы. Также нарушается синтез эстрогенов, что проявляется нарушением эндометриального цикла (аменорея) и слабым развитием женских вторичных половых признаков.
Диагноз может быть поставлен при отсутствии телец Барра в соскобах буккального эпителия у лиц, имеющих женский фенотип или при анализе кариотипа.
Синдром ХХХ ("суперженщины”) — присутствие третьей Х-хромосомы у женщин. Большинство пациентов являются нормальными. У некоторых наблюдаются нарушения умственного развития, нарушения менструального цикла и снижение фертильности (плодовитости).
Синдром ХYY — присутствие лишней Y-хромосомы у мужчин. Большинство пациентов нормальные. У некоторых может наблюдаться агрессивность поведения и легкое отставание в умственном развитии, распространенность — 1:1000 живорожденных мальчиков.
Синдром нестабильности Х-хромосомы — при этом синдроме наблюдается отставание в умственном развитии у 80% мужчин, имеющих поврежденную Х-хромосому, а у женщин — только30% (вероятнее всего, в результате предпочтительной инактивации поврежденной Х-хромосомы). Аномалия заключается в необычно большом количестве повторений нуклеотидных триплетов на самом конце длинного плеча Х-хромосомы (Хq27). Частота распространения — до 1:1000 у мужчин и 1:2000 — у женщин.
ГЕННЫЕ НАРУШЕНИЯ, ПЕРЕДАЮЩИЕСЯ ПО ЗАКОНАМ МЕНДЕЛЯ
Генные пороки могут наследоваться по:
аутосомно-рецессивному типу (порок возникает при условии, что мутантный ген унаследован от отца и матери, которые могут быть здоровы);
аутосомно-доминантному типу (мутантный ген передается от одного из родителей, который страдает аналогичным пороком).
Аутосомные доминантные болезни (табл. 1): большинство аутосомных доминантных заболеваний не являются смертельными, больные доживают до взрослого возраста и могут передавать аномальный ген потомству. Однако иногда наблюдаются спорадические (несемейные) случаи в результате новых мутаций. Характеристиками многих аутосомных болезней являются вариация частоты, с которой аномальный ген проявляется себя клинически в виде болезни (пенетрантность) и степень выраженности этих клинических проявлений у различных индивидуумов (экспрессивность).
Таблица 1
Наиболее важные врожденные заболевания1
|
Доминантный |
Хромосомы2 |
Рецессивный | |
| Семейная гиперхолестеринемия | 19 | 11 | Серповидноклеточная анемия |
| Поликистоз почек (взрослый тип) | 16 | 16 | Альфа-таллассемия |
| Наследственный эллиптоцитоз | 1, 2 | 16 | Бета-таллассемия |
| Нейрофиброматоз (I тип) | 17 | 7 | Кистозный фиброз (муковисцедоз) |
| Наследственный сфероцитоз | 8, 14 | ||
| Болезнь Вон Виллебранда | 12 | 1 | Болезнь Гаучера |
| Синдром Элера-Данлоса | 6 | Семейный гемохроматоз | |
| Опухоль Вильмса | 11 | 7 | Недостаточность миелопероксидазы |
| Семейный полипоз кишечника | 5 | 15 | Болезнь Тау-Сач |
| Ретинобластома | 13 | 14 | ?1-антипротеазная недостаточность |
| Синдром Марфана | 12 | Фенилкетонурия | |
| Ахондроплазия | Альбинизм | ||
| Синдром незавершенного остеогенеза | 17 | 13 | Болезнь Вильсона |
| Острая перемежающаяся порфирия | 1 | Галактоземия | |
| Болезни накопления гликогена | |||
| Хорея Гентингтона | 4 | 22 | Болезнь Хурлера |
| Алкаптонурия | |||
| Врожденная геморрагическая телеангиэктазия | 22 | Метахроматическая лейкодистрофия | |
| Семейный амилоидоз | 18 | ||
Аутосомные рецессивные заболевания: большинство аутосомных рецессивных заболеваний характеризуется недостаточностью ферментов, что приводит к биохимическим дефектам, которые называются врожденной "ошибкой” (нарушением) метаболизма. У людей с гетерозиготным генотипом (носителей) уровень этих энзимов или других нормальных протеинов может быть снижен, но не проявляться в виде болезни (один ген остается функционирующим). При гомозиготном генотипе болезнь развивается в результате недостатка важных белков (например, гемоглобина А при талассемии), присутствия патологических продуктов (например, гемоглобина С при серповидноклеточной анемии) или накопления токсических метаболитов в ферментных системах в результате отсутствия определенных энзимов (например, болезни накопления). При таких заболеваниях больные обычно погибают в детском возрасте, однако в некоторых случаях при соответствующем лечении больные могут доживать до преклонных годов, например, люди с фенилкетонурией могут вести нормальный образ жизни при полном исключении фенилаланина из пищи. Многие аутосомные рецессивные заболевания встречаются часто в семьях с близкородственными браками.
Болезни, передающиеся по связанному с Х-хромосомой типу
Рецессивные1
Красно-зелёная цветовая слепота2
Дефицит глюкозо-6-фосфатдегидрогеназы
Синдром нестабильности Х-хромосомы3
Мышечная дистрофия Даччена
Гемофилия А (дефицит VIII фактора)
Агаммаглобулинемия
Хроническая гранулематозная болезнь
Иммунодефицит Брутона
Тестикулярная феминизация
Синдром Леш-Нихана
Болезнь Фабри
Болезнь Кристмаса (дефицит IX фактора)
Синдром Хантера
Доминантные
Гипофосфатемический рахит (витамин-D-резистентный)
Псевдогиперпаратиреоидизм
1 Упорядочено по частоте встречаемости (синдром нестабильности Х-хромосомы 1:1000, синдром Хантера 1:100000).
2 Полная цветовая слепота (аутосомно-рецессивный тип наследования) встречается очень редко).
3 Синдром нестабильности Х-хромосомы имеет следующую частоту встречаемости: 1:1000 у мужчин и 1:2000 у женщин.
ВРОЖДЕННЫЕ НАРУШЕНИЯ МЕТАБОЛИЗМА
Эти заболевания возникают в результате врожденного нарушения генов, кодирующих синтез определенных энзимов, что приводит к блокированию метаболических путей на определенном этапе. Недостаточность ферментов проявляется чаще в нарушении обмена аминокислот, липидов, углеводов и мукополисахаридов с накоплением субстратов и недостатком продукта ферментных реакций. Это может стать причиной повреждения клеток. Все эти заболевания редки. Большинство имеет аутосомно-рецессивный тип наследования, а некоторые — рецессивные, связанные с Х-хромосомой.
Нарушения обмена аминокислот
Типичным примером являются нарушения обмена фенилаланина и тирозина (рис. 1). При фенилкетонурии отсутствие фенилаланингидроксилазы препятствует преобразованию фенилаланина в тирозин. Это приводит к дефициту тирозина в клетках (с нарушением синтеза меланина и отсутствием пигментации) и в такой же степени — накоплению фенилаланина, который является токсичным для нервных клеток (что приводит к развитию умственной недостаточности). Фенилкетонурия — это пример биохимического нарушения, которое не имеет специфических морфологических признаков в пораженных клетках. Диагноз ставится при нахождении высоких уровней фенилаланина в моче и сыворотке крови. Лечение заключается в исключении фенилаланина из рациона.
Таблица 3
Примеры врожденной недостаточности ферментов, принимающих участие в обмене аминокислот1
Заболевание
Аминокислота
Дефицит фермента
Клинические характеристики
|
Заболевание |
Аминокислота |
Дефицит фермента |
Клинические характеристики |
| Фенилкетонурия | Фенилаланин | Фенилаланин-гидроксилиза | Умеренное недоразвитие, специфический запах изо рта, экзема, повышение уровня фенилаланина в крови. |
| Врожденная тирозинемия | Тирозин | Оксидаза гидроксифенил- пировиноградной кислоты |
Цирроз печени, нарушение функции почечных канальцев; повышение уровня тирозина в крови. |
| Гистидинемия | Гистидин | Гистидиназа | Умственное недоразвитие, нарушения речи. |
| Болезнь мочи в виде кленового сиропа (кетоаминоацидурия) | Лейцин, валин, изолейцин | Оксидаза кетокислот | Умственное недоразвитие, моча с запахом клепового сиропа. |
| Гомоцистинурия | Метионин, гомоцистеин | Цистатионинсинтетаза | Умственное недоразвитие, тромбэмболическая пневмония, эктопия хрусталика. |
1 Все приведенные в этой таблице заболевания имеют аутосомно-рецессивный тип наследования.
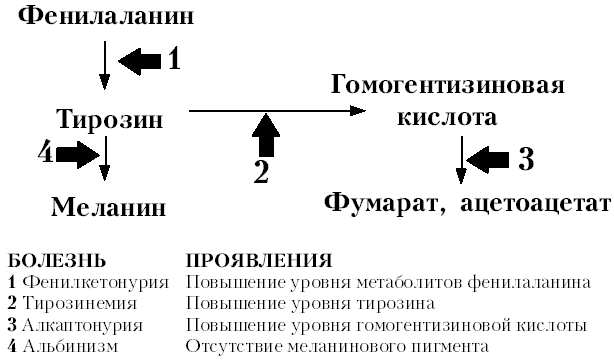
Рис.1. Врожденные нарушения метаболизма фенилаланина и тирозина
Нарушения обмена липидов (болезни накопления липидов)
Недостаток ферментов приводит к нарушению родственных путей метаболизма сфинголипидов. Эта недостаточность приводит к накоплению больших количеств сложных липидов в клетках. Большинство этих энзимов является лизосомными и аномальные липиды накапливаются во вторичных лизосомах, поэтому они еще называются лизосомными болезнями накоплениями. За исключением болезни Фабри, которая является рецессивной и связанной с Х-хромосомой, все остальные являются аутосомно-рецессивными.
Лизосомальные (или липидные) болезни накопления
|
Заболевание |
Дефектный энзим |
Накапливающийся липид |
Поражаемые ткани |
| Болезнь Тау-Сач | Гексозаминидаза А | GM2 ганглиозид | Мозг, сетчатка глаза. |
| Болезнь Гаучара | ?-глюкозидаза(глюкоцереброзидаза) | Глюкоцереброзиды | Печень, селезёнка, костный мозг, головной мозг. |
| Болезнь Ниманна-Пика | Сфингомиелиназа | Сфингомиелин | Головной мозг, печень, селезёнка. |
| Метахроматическая лейкодистрофия | Арилсульфатаза А | Сульфатиды | Головной мозг, почки, печень, периферические нервы. |
| Болезнь Фабри | ?-галактозидаза | Тригексозид церамида | Кожа, почки. |
| Болезнь Краббе | Галактозилцерамид | Галактоцереброзид | Головной мозг. |
Накопление липидов наблюдается в различных клетках в зависимости от варианта заболевания. Поражение паренхиматозных клеток приводит к развитию дистрофий и некроза этих клеток. Поражение нейронов, например, при болезни Тау-Сач и детских формах болезней Гаучера и Ниманна-Пика, вызывает тяжелые нарушения умственного развития и смерть. Почечная недостаточность наблюдается при ренальной форме болезни Фабри. Более легкие формы болезней Гаучера и Ниманна-Пика у взрослых приводят к накоплению липидов в ретикулоэндотелиальных клетках, что сопровождается увеличением печени и селезенки.
Диагностика:
Клинические проявления — при болезни Тау-Сач липиды накапливаются в сосочке зрительного нерва, что приводит к появлению вишнево-красной метки, видимой при офтальмоскопии. При болезни Фабри наблюдается диффузное поражение кожи, а при болезни Гаучера — гепатоспленомегалия.
Микроскопическое исследование — при световой микроскопии пораженных тканей, таких как мозг, костный мозг, печень и селезенка, определяются аномальные клетки, увеличенные в объеме за счет накопления большого количества липидов в цитоплазме. Пораженные клетки при болезни Тау-Сач и Ниманна-Пика имеют пенистую цитоплазму. При болезни Гаучера клетки имеют характерную исчерченность цитоплазмы ("гофрированная бумага”).
При электронной микроскопии характерные включения представляют собой перерастянутые лизосомы. При болезни Тау-Сач они завитые, при болезни Ниманна-Пика они имеют вид параллельных пластинок ("тельца-зебры”), при болезни Гаучера накапливающиеся липиды упорядочены в виде поленицы дров.
Нарушения обмена гликогена
Болезни накопления гликогена связаны с недостатком ферментов, участвующих в обмене гликогена. Большинство этих болезней передается аутосомно-рецессивно и проявляются в младенческом или детском возрасте.
Болезни накопления гликогена
|
Тип |
Дефектный энзим |
Тяжесть |
Поражаемые ткани |
| I (болезнь Гирке) | Глюкозо-6-фосфатаза | Тяжелая | Печень, почки, кишечник. |
| II (болезнь Помпе) | ?-1,4-глюкозидаза | Летальная | Распространённое поражение, но больше всего - сердце. |
| III (болезнь Кори) | Амило-1,6-глюкозидаза (деполимеризующий) | Умеренная | Распространённое поражение, но больше всего - печень. |
| IV (болезнь Андерсена) | Амило-1,4>1,6-трансглюкозидаза (полимеризующий) | Летальная | Распространённое поражение, но больше всего - печень. |
| V (болезнь Мак Ардля) | Мышечная фосфорилаза | Умеренная | Скелетная мускулатура |
| VI (болезнь Херса) | Печеночная фосфорилаза | Умеренная | Печень |
| VII-XII | Очень редкие болезни | Различная | Различные |
Гликоген накапливается в цитоплазме и может быть выявлен при электронной микроскопии в виде гранул. При обычной фиксации срезов гликоген вымывается раствором формалина. Пораженные клетки на обычных гистологических срезах при световой микроскопии выглядят пустыми. Для определения гликогена в клетках необходима фиксация в этиловом спирте и окраска кармином Беста или ШИК-реакция.
Поражение печени проявляется в виде гепатомегалии, фиброза и печеночной недостаточности; поражение сердца приводит к сердечной недостаточности. Поражение печени, например, при 1 типе, проявляется гипогликемией, так как основным источником глюкозы крови является расщепленный гликоген печени. Нарушение доставки глюкозы к скелетным мышцам проявляется судорогами и слабостью.
Нарушения обмена мукополисахаридов (мукополисахаридозы)
Мукополисахаридозы являются редкими врожденными лизосомными болезнями накопления, при которых недостаток лизосомных ферментов приводит к накоплению мукополисахаридов (гликозаминогликанов) в лизосомах различных клеток. Все они передаются аутосомно-рецессивно, за исключением синдрома Хантера, который передается рецессивно, связанно с Х-хромосомой.
Мукополисахаридозы
|
Тип |
Дефектный энзим |
Накапливающиеся МПС |
Поражаемые ткани |
Тип наследования |
Тяжесть |
| I (синдром Херлера) | ?-L-идуронидаза | Гепарансульфат, дерматансульфат | Кожа, кости, роговица, мозг, печень, селезёнка. | Аутосомно- рецессивный | Тяжелая |
| II (синдром Хантера) | L-идуроносульфат сульфатаза | Гепарансульфат, дерматансульфат | Кожа, кости, сердце, уши, сетчатка. | Связанный с Х-хромосомой рецессивный | Средняя |
| III (синдром Санфиллиппо) | Многие | Гепарансульфат | Мозг, кожа. | Аутосомно- рецессивный | Средняя |
| IV (синдром Морквио) | N-ацетилгалактозамин-5- сульфатаза | Кератансульфат, хондроитинсульфат | Кожа, мозг, кости, глаза. | Аутосомно- рецессивный | Легкая |
| V-VII | Редкие заболевания с поражением многих энзимов | Различные | Различные | Аутосомно- рецессивный | Легкая |

